|
Simulation d'un spectre RMN avec le logiciel HNMR Spectrum |
|
|
|
Réaliser un
spectre RMN avec ACD/Labs.
1)- Le logiciel
ACD/ChemSketch (Freeware)
2)- Récupération
des données de la molécule : |
|
|
Réaliser un spectre
RMN avec ACD/Labs.
1)- Le logiciel
ACD/ChemSketch (Freeware)
-
Télécharger le logiciel :
-
Cliquer sur le lien suivant :
https://www.acdlabs.com/resources/free-chemistry-software-apps/
-
On obtient la fenêtre suivante :

-
Cliquer sur
-
Puis créer un compte : Cliquer
sur
![]()
-
Cliquer sur :![]()

-
Puis cliquer sur
![]() .
.
-
Enfin, installer le fichier
-
Cliquer sur l’application :
chemsk12.exe
-
La fenêtre suivante apparaît :
-
Cliquer sur
![]() et suivre la
procédure pour installer le logiciel.
et suivre la
procédure pour installer le logiciel.
-
Une fois le logiciel installé,
cliquer sur le raccourci : 
-
Il apparait la fenêtre suivante :

-
Cliquer sur OK (le logiciel est
en anglais, mais facilement compréhensible)
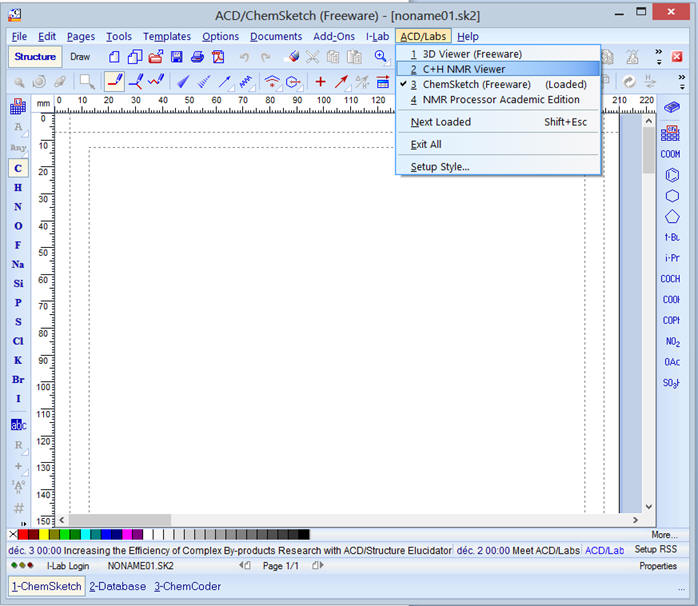
-
Cliquer sur ACD/Labs
-
Dans le menu déroulant, cliquer
sur
![]()
-
Il apparaît la fenêtre suivante :
-
Sélectionner :
![]()
-
Dans le menu : Tools,
sélectionner Simulation
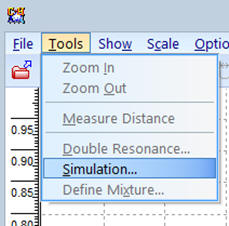
-
Il apparaît la fenêtre suivante :

-
Pour pouvoir construire le
spectre RMN de la molécule, il faut connaître les
différents déplacements chimiques des groupes de protons
équivalents qui constituent la molécule.
-
La molécule ne doit pas être trop
complexe (moins de 5 groupes de protons équivalents)

2)-
Récupération des données de la molécule :
-
Site qui permet d’accéder aux
données :
-
https://sdbs.db.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi
-
Cliquer
sur : I agree the disclaimer and use SDBS
-
Il apparaît la page suivante :
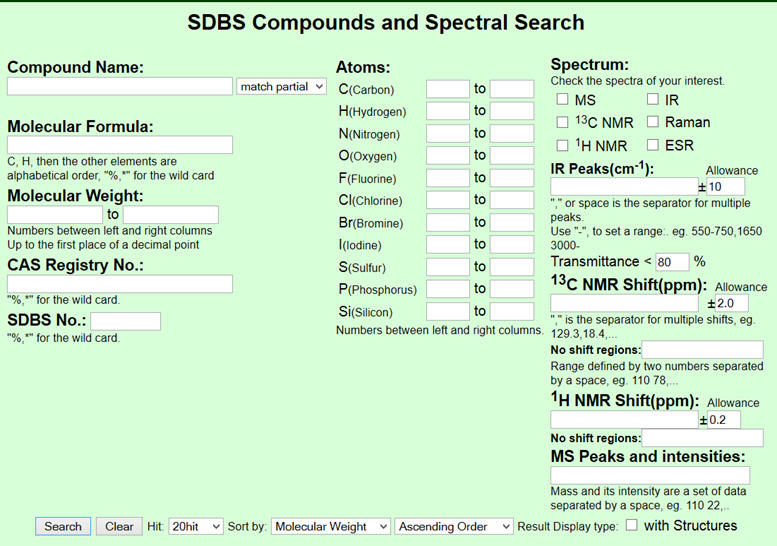
-
On peut entrer la formule
moléculaire.
-
Pour exemple on choisit
l’éthanol : C2H6O

-
On demande d’afficher le
spectre
1H NMR (on peut cocher d’autres spectres)
-
On clique sur « Search ».

-
On clique sur le lien : 1300 (il
s’agit bien de l’éthanol)
-
Puis pour afficher le spectre
RMN
de la molécule, on clique sur le lien :
-
1H
NMR : 90 MHz in CDCl3
-
On obtient le spectre suivant
avec les données :

3)-
Simulation du spectre avec le logiciel HNMR Spectrum.
-
Il faut entrer les différentes
valeurs dans la page suivante :
-
On entre la valeur de la
fréquence : f = 90 MHz (on choisit la même valeur que
celle donnée par le site)
-
Cette fréquence
f
représente la valeur de la fréquence de résonance des protons de
l’échantillon de référence.
-
Elle est liée à la valeur du
champ magnétique imposé par l’appareil de RMN.
-
Ainsi un appareil de RMN à 9,4 T
est appelé spectromètre de 400 MHz.
-
Il y a proportionnalité entre la
valeur de l’intensité du champ magnétique appliqué et la
fréquence f de résonance des protons de l’échantillon de
référence.
►
Tableau de correspondance :
|
Valeur de l’intensité
du champ magnétique
de l’appareil RMN |
1,41 T |
2,11 T |
2,4 T |
4,7 T |
7,1 T |
9,4 T |
|
Fréquence de
résonance des protons
de l’échantillon
de référence |
60 MHz |
90 MHz |
100 MHz |
200 MHz |
300 MHz |
400 MHz |
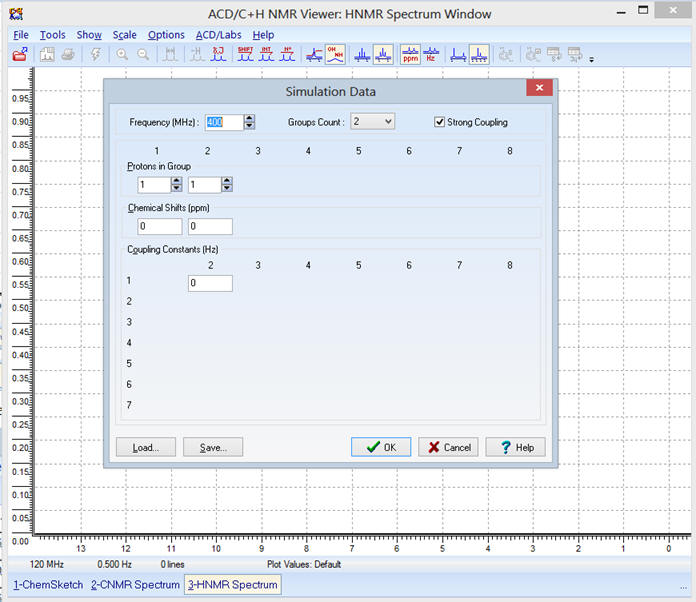
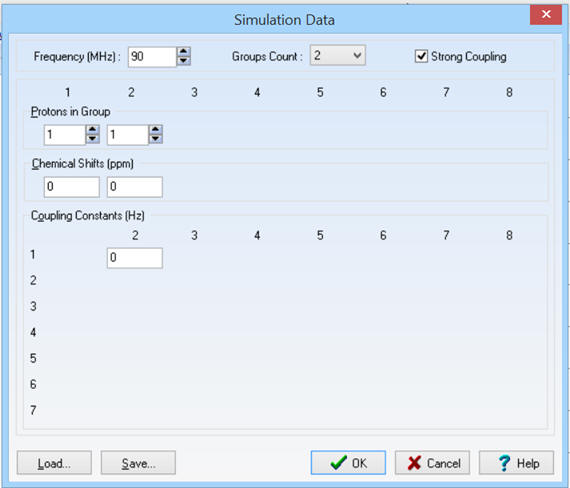
-
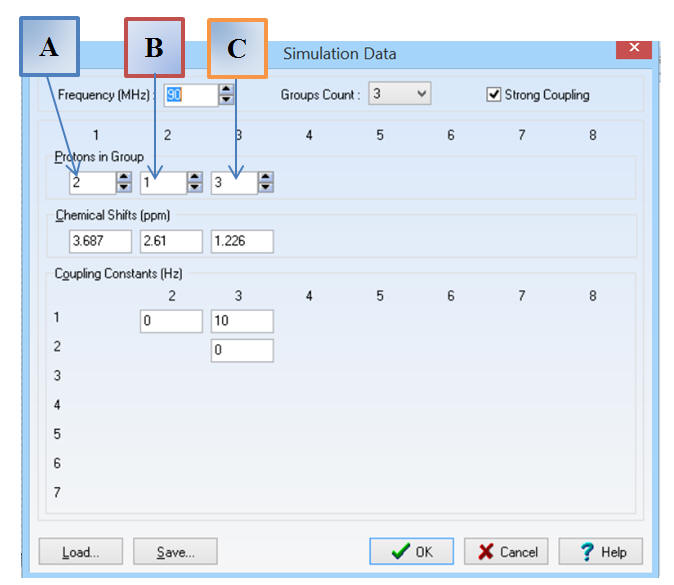
Il y a trois groupes de protons
équivalents : Groupe Count : 3
-
Il
apparaît la fenêtre suivante :

-
On indique le nombre de protons
par groupe et le déplacement chimique correspondant :
-
On respecte l’ordre
A,
B et
C.
-
Maintenant, il faut entrer les
valeurs des constantes de couplage :
- Dans le cas présent, les constantes de couplage ne sont pas données.

-
Valeur de la constante de couplage :
-
Constante de couplage J entre
les deux protons :
Distance entre les 2 pics d’un signal.
-
Les constantes de couplage
J sont exprimées en hertz.
-
La valeur de la constante de couplage est indépendante de
l’intensité du champ magnétique appliqué et ne dépend que du
nombre et de la nature des liaisons séparant les deux protons et
de la disposition spatiale des protons.
►
On utilise les règles suivantes :
-
Les effets du couplage
disparaissent rapidement avec la distance.
-
Alors que les atomes d'hydrogène
portés par des atomes adjacents de carbone présentent
un effet de couplage notable (J = de 6 Hz à 8 Hz),
les atomes d'hydrogène éloignés (deux hydrogènes séparés par
plus de deux carbones) les uns des autres ne subissent presque
pas d’effets réciproques (J = de 0 Hz à 1 Hz).
-
Par exemple, dans le spectre de
protons de l'éthanol, le groupe CH3 est divisé en un
triplet avec un rapport d'intensité de 1:2:1 par les deux
protons voisins du groupe CH2.
-
De même, le groupe
CH2
est divisé en un quartet avec un ratio d'intensité de 1:3:3:1
par les trois protons voisins du groupe CH3.
-
En principe, les deux protons du
groupe CH2 devraient être également divisés à nouveau
en un doublet pour former un doublet de quartets à cause du
couplage avec le proton hydroxyle, mais l'échange
intermoléculaire du proton hydroxyle acide entraîne l'annulation
de ce couplage.
-
Le pic du proton hydroxyle
apparaît généralement sous la forme d'un singulet.
-
Du fait de la mobilité de l'atome
d'hydrogène du groupe hydroxyle, le couplage avec les autres
protons (protons portés par le carbone voisin) disparaît.
-
On prend :
JAC
= J13 = 7 Hz ou 10 Hz
-
Pour
JAB =
J12
= 0 Hz et JBC = J23 = 0 Hz

-
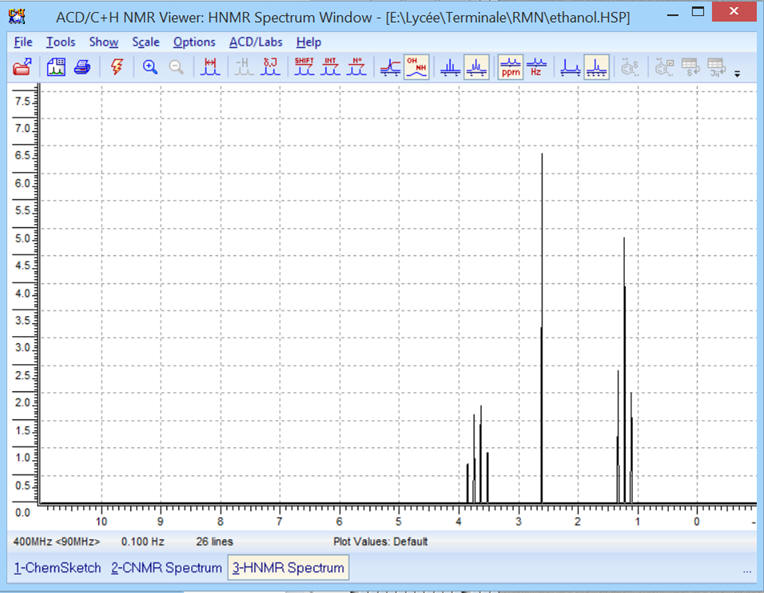
On obtient le spectre RMN
suivant :
-
Avec une fréquence de 400 MHz, on
obtient le spectre RMN suivant :

-
Le spectre est plus resserré.
-
On peut Zoomer :

-
On peut faire apparaître la
courbe d’intégration :

|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
-
Ensemble des outils :
►
![]() Permet
de zoomer :
Permet
de zoomer :
-
On sélectionne la partie du
spectre que l’on veut étudier tout en maintenant le clic gauche
de la souris, puis on relâche :

-
On obtient le spectre suivant :
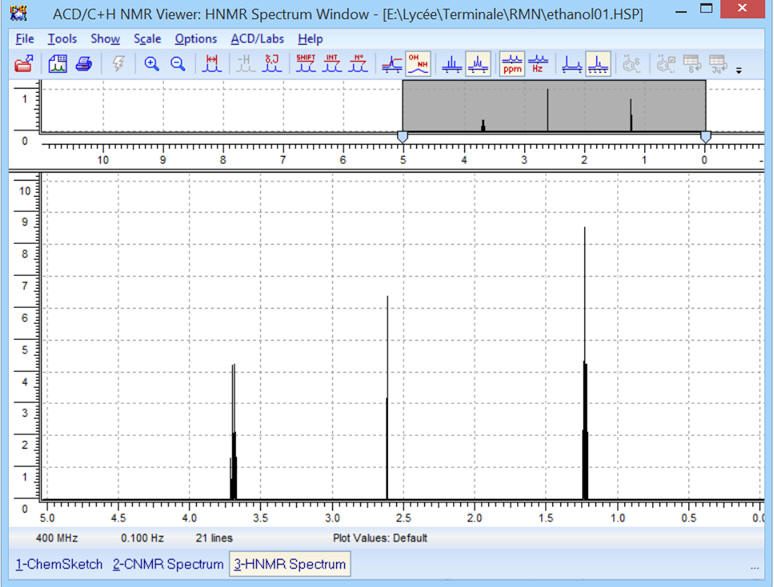
![]()
►
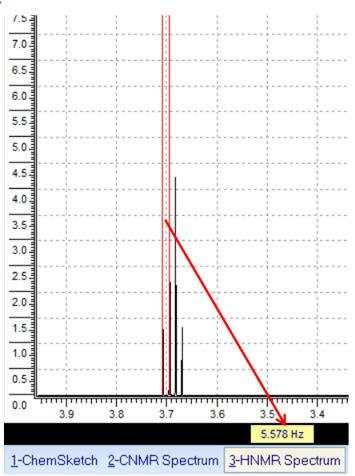
![]() Mesure
de distance : permet d’effectuer des mesures de distance en Hz
(constante de couplage, ...).
Mesure
de distance : permet d’effectuer des mesures de distance en Hz
(constante de couplage, ...).
-
Exemple :
![]()
►
![]() :
Simulation de spectre
HRMN :
:
Simulation de spectre
HRMN :
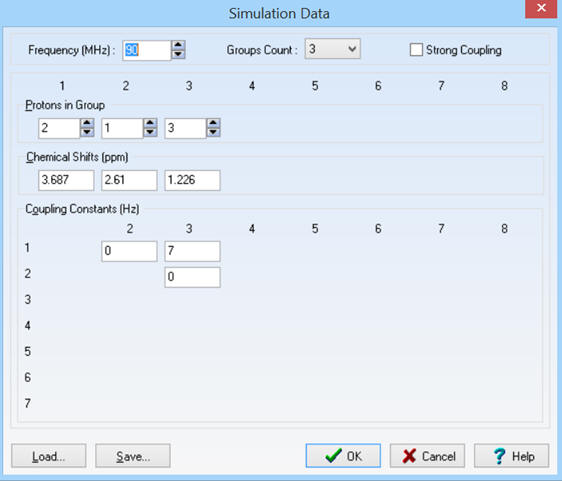
-
Permet de rentrer les différentes
valeurs pour simuler un spectre RMN.
-
Exemple : Pour la molécule
d’éthanol :
-
Il faut indiquer :
-
Nombre de groupes : 3
-
Nombre de protons dans chaque
groupe : ![]()
-
Constantes de couplage :

![]()
►
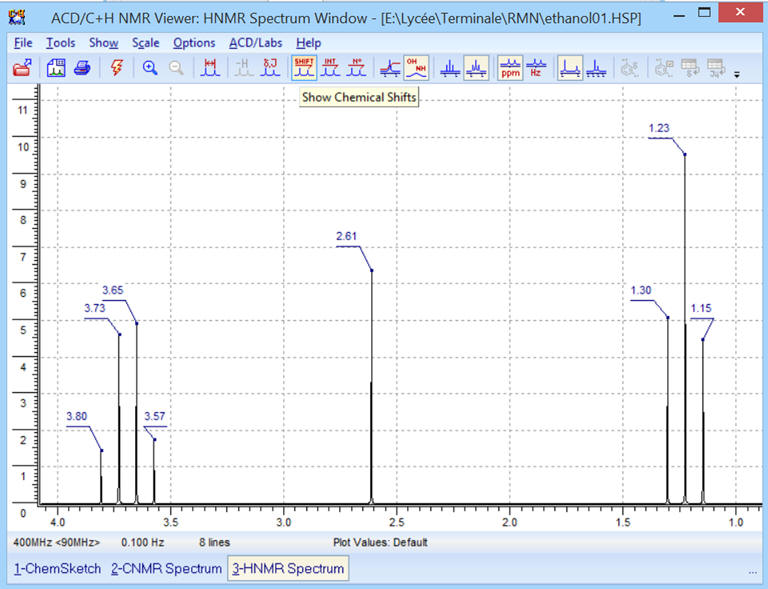
![]() Affichage
des déplacements chimiques de chaque pic des différents signaux.
Affichage
des déplacements chimiques de chaque pic des différents signaux.
-
Exemple : Pour la molécule
d’éthanol :
![]()
►
![]() Affiche
l’intensité de chaque pic des différents signaux.
Affiche
l’intensité de chaque pic des différents signaux.
-
Exemple : Pour la molécule
d’éthanol :

![]()
► ![]() Affiche
le numéro de groupe de chaque pic de chaque signal.
Affiche
le numéro de groupe de chaque pic de chaque signal.
-
Exemple : Pour la molécule
d’éthanol :

![]()
►
![]() Affiche
la courbe d’intégration.
Affiche
la courbe d’intégration.
-
Exemple : Pour la molécule
d’éthanol :

![]()
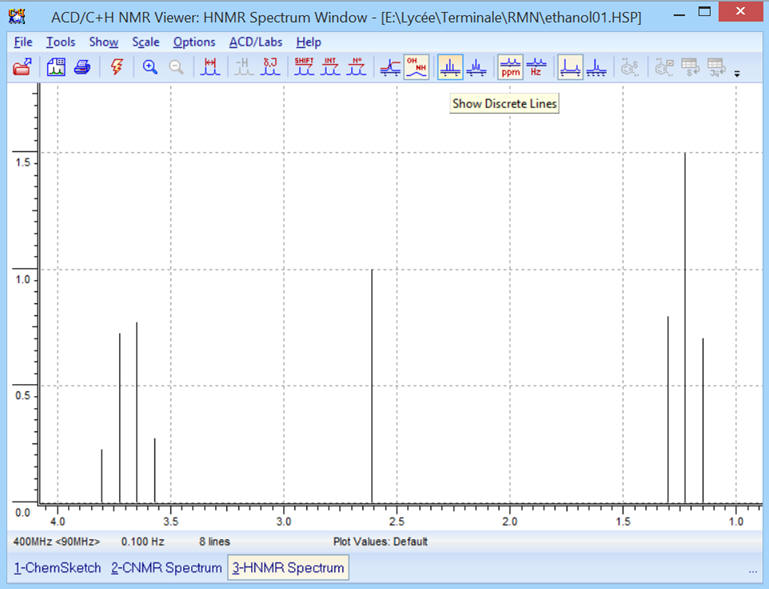
► ![]() Affichage
avec des lignes simples:
Affichage
avec des lignes simples:
-
Le spectre ne fait apparaître que
des lignes.
![]()
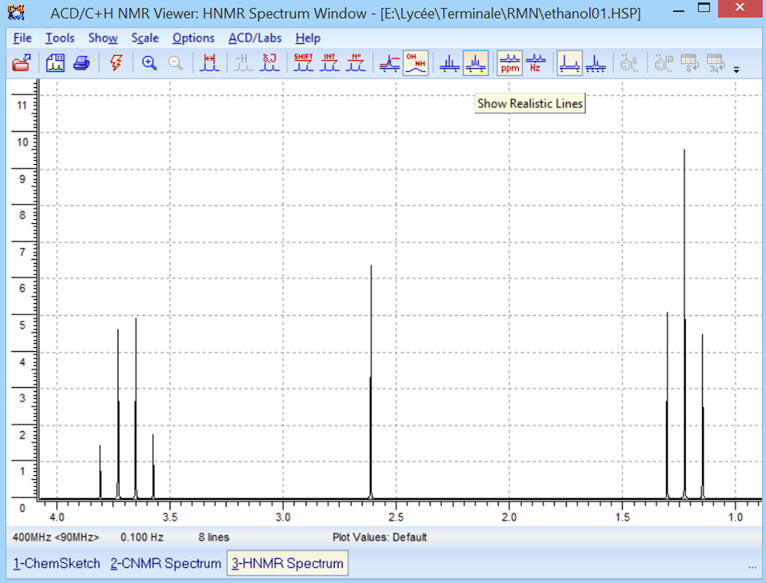
► ![]() Affichage
des lignes plus proche de la réalité (plus réaliste).
Affichage
des lignes plus proche de la réalité (plus réaliste).
![]()
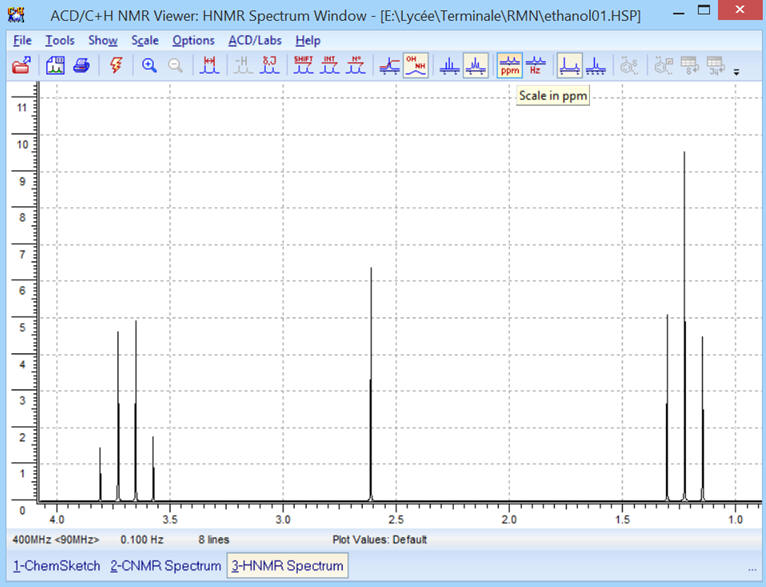
►
![]() Affichage
des déplacements chimiques en ppm
Affichage
des déplacements chimiques en ppm
![]()
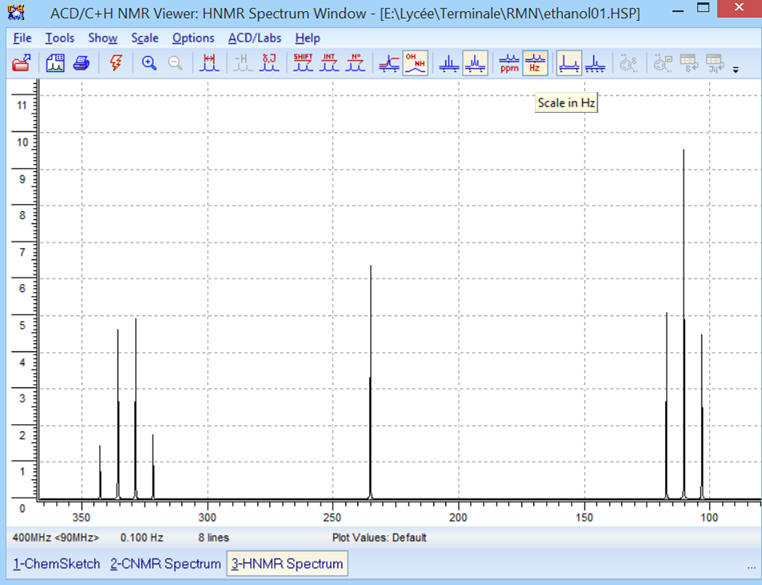
► ![]() Affichage
des déplacements chimiques en Hz.
Affichage
des déplacements chimiques en Hz.
![]()
► ![]() Affichage avec les échelles les
mieux adaptées.
Affichage avec les échelles les
mieux adaptées.
-
Adapte le spectre RMN à la
fenêtre.
-
Permet d’obtenir l’affichage le
plus lisible, le zoom le mieux adapté.

![]()
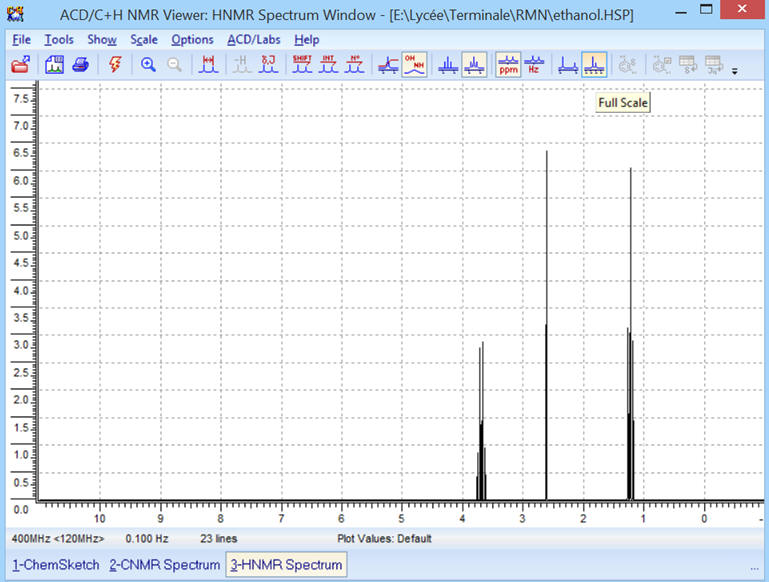
► ![]() Affichage
avec les échelles complètes.
Affichage
avec les échelles complètes.
![]()
-
Dans le menu « Options » :
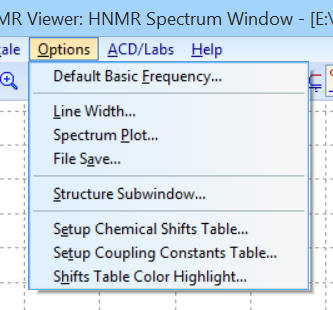
-
On peut choisir l’intervalle de
spectre : les valeurs par défaut

![]()
|
|